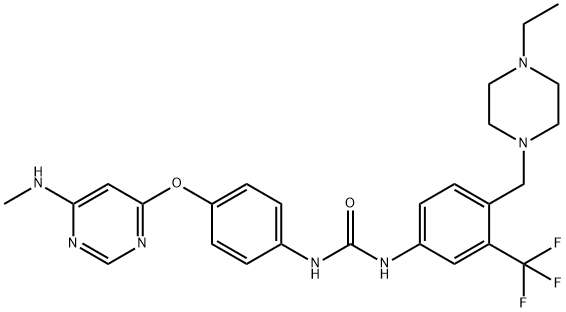
中文同义词:
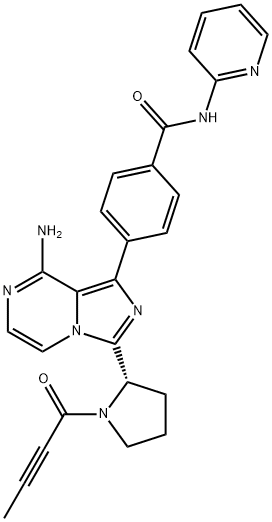
4-[8-氨基-3-[(2S)-1-(1-氧代-2-丁炔-1-基)-2-吡咯烷基]咪唑并[1,5-A]吡嗪-1-基]-N-2-吡啶基-苯甲酰胺;阿卡拉替尼;艾克替尼;阿卡卢替尼;阿可替尼;苯甲酰胺, 4-[8-氨基-3-[(2S)-1-(1-氧代-2-丁炔-1-基)-2-吡咯烷基]咪唑并[1,5-A]吡嗪-1-基]-N-2-吡啶基-;阿卡替尼;BTK抑制剂(ACALABRUTINIB)
英文同义词:
ACP196;ACP-196;acalabrutinib;Benzamide, 4-[8-amino-3-[(2S)-1-(1-oxo-2-butyn-1-yl)-2-pyrrolidinyl]imidazo[1,5-a]pyrazin-1-yl]-N-2-pyridinyl-;ACP196,Acalabrutinib;4-[8-Amino-3-[(2S)-1-(1-oxo-2-butyn-1-yl)-2-pyrrolidinyl]imidazo[1,5-a]pyrazin-1-yl]-N-2-pyridinylbenzamide;EOS-60753;(S)-4-(8-amino-3-(1-but-2-ynoylpyrrolidin-2-yl)imidazo[1,5-a]pyrazin-1-yl)-N-(pyridin-2-yl)benzamide
相关类别:
细胞生物学试剂;医药原料;医用原料;原料药;医药原料药;第二代BTK抑制剂;医用原料;原料药;化工原料
密度
1.37±0.1 g/cm3(Predicted)
酸度系数(pKa)
11.47±0.70(Predicted)
简介
Acalabrutinib(ACP-196)是选择性的第二代BTK抑制剂,抑制B细胞表面原抗体信号通路的激活。它具有很好的靶标特异性,对BTK的选择性比对其他TEC激酶家族成员如ITK、TXK、BMK和TEC的选择性高323-, 94-, 19-, 9-倍。对EGFR没有活性。
用途
ACP-196是一种实验性抗癌药物,是一种选择性的Bruton酪氨酸激酶(BTK)抑制剂。该激酶传输来自B细胞受体(BCR)的信号,因此任何遗传性BTK突变都会导致B细胞免疫缺陷。因此,靶向B细胞信号传导的BTK抑制剂已显示出治疗慢性淋巴细胞性白血病(CLL)的巨大希望。
生物活性
体外:ACP-196(Acalabrutinib)在原代人CLL细胞的体外信号传导测定中抑制ERK,IκB和AKT下游靶标的酪氨酸磷酸化。
体内:在人CLL NSG异种移植模型中,ACP-196显示了靶向作用,包括PLCγ2,ERK的磷酸化降低和CLL细胞增殖的显着抑制。ACP-196显着降低小鼠脾脏中的肿瘤负荷。在TCL1过继转移模型中,ACP-196处理降低BTK,PLCγ2和S6的磷酸化。最值得注意的是,与接受载体的小鼠相比,acalabrutinib导致存活率显着增加。 ACP-196(每天两次100毫克)评估小鼠受损小动脉的血栓形成,表现出更强的抑制BTK选择性,几乎没有抑制血小板活性。
靶点
Target Value
BTK
(in a human whole-blood CD69 B cell activation assay) 3nM
BTK
(in a human whole-blood CD69 B cell activation assay)
3nM
体外研究
在初级人源慢性淋巴细胞白血病细胞的体外信号检测中,Acalabrutinib抑制下游靶标ERK、IKB、AKT的酪氨酸磷酸化。在9种与BTK半胱氨酸所在位置一致的激酶中,Acalabrutinib对BTK的选择性高于对其他激酶。Acalabrutinib不抑制EGFR、ITK和TEC,对EGFR在Y1068和Y1173位点的磷酸化没有影响。相对于ibrutinib,Acalabrutinib具有更高的IC50值,并几乎对 ITK, EGFR, ERBB2, ERBB4, JAK3, BLK, FGR, FYN, HCK, LCK, LYN, SRC以及YES1的激酶活性没有抑制作用。
体内研究
对小鼠进行ACP-196的口服处理,在CD19+脾细胞中抑制了anti-IgM诱导的CD86表达,此抑制作用为剂量依赖性,ED50为0.34 mg/kg。在处理后3h,ACP-196抑制了>90%的CD86表达水平。
 15623309010
15623309010







 点击咨询官方QQ客服
点击咨询官方QQ客服 微信号:15623309010
微信号:15623309010 手机号:15623309010
手机号:15623309010 点击咨询官方在线客服
点击咨询官方在线客服