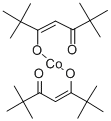
双(2,2,6,6,-四甲基-3,5-庚二酮酸)钴 时间:2026-07-16 09:47 作者:system 点击:次 化学性质 熔点143°C 形态粉末 水溶解性Soluble in hydrocarbons, alcohols and ethers. Slightly soluble in water. CAS 数据库13986-53-3 用途与合成方法 上一篇:氯代三叔丁基磷化金(I) 下一篇:三苯基六氯锑酸碳